

 补体调节分子异常
补体调节分子异常
-
补体系统是人类固有免疫系统的重要组成成分,广泛参与机体抗病原微生物的防御反应和免疫调节,也可导致免疫病理损伤。补体系统由补体固有成分、补体调节因子和补体受体等30余种蛋白组成。激活补体系统的途径有3条,分别为经典途径、凝集素途径及旁路途径。经典途径始于免疫复合物与C1q的结合,依次激活C1r和C1s,剪切C4和C2形成经典途径C3转化酶C4b2b。凝集素途径始于病原体表面的甘露糖与甘露糖结合凝集素 (mannose-binding lectin, MBL) 的结合,活化与MBL结合的丝氨酸蛋白酶MASP,剪切C4和C2形成C3转化酶。旁路途经始于C3分子自发裂解产生的C3b,C3b与B因子结合后在P因子及D因子的作用下可形成旁路途径C3转化酶C3bBb。三条通路在C3转化酶阶段汇合。C3转化酶可裂解C3,产生C3b,形成C5转化酶 (经典与凝集素途径为C4b2b3b,旁路途径为C3bnBb),进一步裂解与C3b结合的C5产生C3b5b。C3b5b参与补体活化的终末阶段,依次与C6、C7、C8及C9结合,最终形成膜攻复合体 (membrane attack complexes, MACs),造成靶细胞的溶解和破裂。正常状态下,补体系统在严密调控机制下发挥其生物学功能,而当补体调节出现异常时,可导致宿主细胞的损伤。
目前认为,aHUS的主要发病机制为补体过度激活造成了内皮细胞的损伤。其中旁路途径的过度激活在aHUS发病机制中的重要地位已得到公认。旁路途径过度激活的原因主要为补体调节因子异常,包括H因子 (complement factor H, CFH)、I因子 (complement factor I, CFI) 和膜辅助蛋白 (membrane cofactor protein, MCP)。这些补体调节因子的主要生物学功能为抑制补体在宿主细胞表面的异常激活。因此,上述因子的基因缺陷或自身抗体的产生均可导致补体异常激活以及宿主细胞的损伤。由于肾小球内皮细胞的位置及其自身的结构,使其成为易受损的靶细胞。除补体调节因子外,补体固有成分C3及B因子的异常在HUS中也有报道。aHUS患者中,约有一半人具有某种补体分子的基因缺陷。
一、H因子
CFH是一种主要由肝脏合成的糖蛋白,由20个短一致重复片段(short consensus repeats, SCRs)结构域组成。CFH是补体系统,特别是补体旁路激活途径的一种重要的负性调节因子。其生物学功能主要是抑制循环中和细胞表面补体旁路途径的激活,具体包括:1.与B因子竞争结合C3b,抑制旁路途径C3转化酶的形成。2.从已形成的旁路途径C3转化酶中置换出C3b,加速旁路途经C3转化酶的灭活。3.作为CFI的辅助因子, 加速CFI灭活补体三条途径产生的C3b。CFH上有很多与其他分子的结合位点,主要包括与肝素,C3b,CRP,唾液酸,C3d等分子的结合位点。这些结合位点的分布使CFH上形成两个重要的功能区:N端功能区主要在液相中发挥补体抑制作用, C端功能区主要具有与细胞表面结合的作用。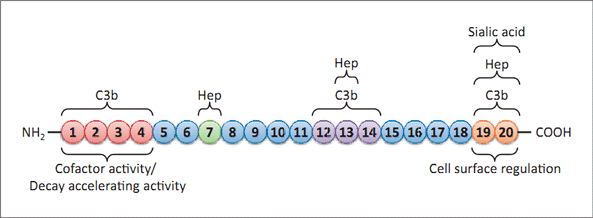
图 H因子结构(引自2010, Nephron Clinical Practice )
CFH浓度降低或功能障碍均可导致其对宿主细胞的保护能力下降。当补体激活造成毛细血管内皮细胞受损后,可发生内皮下基质暴露,补体系统及凝血系统被进一步激活,最终导致血小板消耗、微血栓形成以及红细胞破坏。CFH基因缺陷或循环中抗CFH自身抗体产生均可导致其浓度降低或功能障碍。
CFH基因位于1号染色体长臂3区2带,这个区域是补体活化调节因子基因簇的所在区, 编码CFH的基因与编码衰变加速因子、补体受体、MCP、C4结合蛋白 (C4 binding protein, C4BP)、凝固因子XⅢb等的基因相邻。aHUS中有6-11%是由于基因缺陷造成的。aHUS患者中60-80%的CFH基因缺陷与C端功能缺失相关,主要影响了CFH与细胞表面结合的能力。CFH水平下降不仅可以存在于aHUS患者中,也存在于无症状的患者亲属。目前发现的大多数CFH基因缺陷呈常染色体显性遗传,具有不同的外显率。动物实验也已表明HUS发病机制主要为CFH C端功能缺陷而非CFH功能完全丧失。当CFH基因纯和缺陷时,CFH水平将出现明显下降,但杂合突变的患者体内,CFH水平可出现降低、正常甚至增高。
CFH水平正常不能排除其功能受损。CFH自身抗体存在时,可导致CFH水平正常但活性降低。CFH自身抗体可造成其与配体的结合能力下降,从而引起功能障碍。CFH自身抗体多见于青少年患者。
二、I因子
CFI是另一种由肝脏合成的补体调节因子,是分别由一条重链与轻链组成的二聚体,主要在循环(液相)中发挥作用。其生物学功能是通过降解C3b及C4b而抑制旁路途径C3转化酶的形成,从而抑制补体的激活。CFI生物学功能的发挥依赖于与辅助因子CFH、C4BP及MCP的相互作用。
CFI基因位于4号染色体长臂2区5带。CFI基因缺陷外显率较低,因此大多为散发病例而非家族遗传。CFI基因缺陷时,补体旁路途径不受控制,其结果类似于CFH基因缺陷,最终会导致TMA的发生。
三、 膜辅助蛋白
MCP又称为CD46,是一种广泛表达于细胞表面的跨膜补体调节因子。除红细胞外,MCP几乎表达于体内的所有细胞。其生物学功能为辅助CFI降解沉积于细胞表面的C3b和C4b。其编码基因毗邻CFH编码基因,结构基本单位也为SCR结构域。
与CFH基因突变相似,MCP基因缺陷可导致其表达量减少、与C3b结合能力降低及CFI辅助活性降低,引起补体在细胞表面的过度激活。MCP基因缺陷可以常染色体显性或常染色体隐性方式遗传。但单纯MCP基因缺陷并不一定致病,携带MCP基因缺陷患者病情较轻,这可能与其他因素的参与有关。
四、B因子
五、其他
血栓调节蛋白 (Thrombomodulin, TM) 基因缺陷可引发aHUS。TM是一种普遍存在于内皮细胞表面的糖蛋白,具有抗凝、抗炎和细胞保护的作用。其可在辅助因子 (CFH和C4BP) 存在的条件下辅助CFI降解C3b,还可激活羧肽酶原B,加速过敏毒素C3a和C5a的降解。蛋白C具有抑制血栓形成的作用,TM还可激活蛋白C,从而发挥其抗凝及促纤溶的作用。TM基因缺陷可影响其与配体的结合,从而影响其对补体的调节功能。