

 ADAMTS13或vWF异常
ADAMTS13或vWF异常
-
血管性血友病因子 (von Willebrand factor, vWF) 及其裂解酶ADAMTS-13 (A Disintegrin-like And Metalloproteinase with ThromboSpondintype-1 motif; number 13) 在TTP的发病机制中占有重要地位。
vWF合成于血管内皮细胞、巨核细胞及血小板中。vWF单体 (约275KD)在内质网中合成后,通过在其分子羧基端形成二硫键,形成二聚体,并进一步在高尔基体中形成多聚体,经过糖基化修饰和弗林蛋白酶的剪切,最终形成超大多聚体 (ultra-large, UL-vWF) 贮存于内皮细胞的 Weibel-Palade小体以及巨核细胞或血小板的α颗粒中。正常状态下,内皮细胞可以稳定的分泌少量的vWF。但当内皮细胞受到刺激后(内皮细胞受损或血小板粘附于内皮细胞),大量UL-vWF被分泌,并以线样结构粘附于内皮细胞表面。在血流剪切力的作用下,线样的vWF分子伸展并暴露出酶切位点,可被ADAMTS-13自A2结构域的Tyr1605-Met1606处切割,形成分子量500kDa-20,000kDa 的多聚体。vWF通常以球形多聚体的形式存在于循环中。ADAMTS13对vWF的剪切对于防止微血管中的血栓形成具有重要的意义。
成熟的vWF上有与凝血VIII因子、内皮下胶原、血小板糖蛋白Ibα (GPIα)、干扰素αIIbβIII的结合位点,这些结合位点是vWF发挥生物学功能的基础。在正常血液循环中,vWF以球形多聚体的形式存在,与血小板GPIα的结合位点被封闭,但与内皮下胶原的结合位点始终暴露。当血管内皮受到损伤,内皮下胶原暴露,包括vWF在内的各种黏附分子会在损伤部位聚集,vWF多聚体粘附于胶原,在血流剪切力的作用下其分子结构展开,GPIα结合位点暴露,使血小板停留并黏附于损伤局部的内皮下。vWF与GPIα的结合还可以导致血小板与vWF结合的其他位点 (如GPIIb/IIIa) 大量活化,形成二者相互结合的正反馈,从而在血管损伤局部逐渐形成血小板初级止血栓。血浆中的vWF与VIII因子结合后,可稳定VIII因子,延长其半衰期,内外源性凝血系统被激活,最终形成牢固的次级止血栓。
当血管损伤时,血流中的血小板自身难以抗拒血流剪切力的作用而停留于血管损伤局部,vWF为血小板的黏附聚集提供了介质,使生理性止血过程得以顺利进行。然而,若该反应不能得到有效的生理调控,血小板会不断聚集,血管损伤局部便会形成血栓而非生理性止血。ADAMTS-13便是生理止血过程中重要的“刹车”装置之一。
ADAMTS-13又称为vWF裂解酶,全名为具I型凝血酶敏感蛋白模体的解整体蛋白域和金属蛋白酶域,主要在肝星状细胞中合成,在血管内皮细胞、巨核细胞或血小板中也有合成,在肾脏足细胞中可微量表达。ADAMTS-13主要的生物学功能为裂解vWF。在血流剪切力的作用下,其作用于vWF A2 区 842 酪氨酸 843 蛋氨酸间的肽键,将 vWF 多聚体裂解为大小不等的小分子肽段,在生理状态下调控 vWF 的结构与功能。ADAMTS-13可作用于刚从细胞中分泌的UL-vWF,防止UL-vWF网罗血小板形成病理性血栓。在血管损伤局部,ADAMTS-13剪切vWF,防止在生理性止血过程中在血管损伤局部形成血栓。
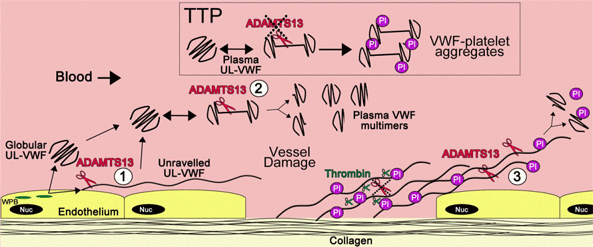
图 TTP中ADAMTS13参与的发病机制。 (引自2011, Blood)
ADAMTS13活性高于正常活性的10%即可防止血小板血栓的形成。但由于ADAMTS13活性受多种因素的影响,临床尚未设定不引发微血管血栓ADAMTS13活性的阈值。
引起ADAMTS132活性降低的因素包括遗传缺陷和免疫因素。由基因缺陷引起的ADAMTS13水平降低或功能不良是先天性TTP病因,而免疫因素引起的抗ADAMTS13抗体形成是特发性TTP的病因。
除ADAMTS13异常外,若vWF因子基因突变引起其分子结构不容易被剪切,也可引发TTP。